

Wat zijn sequencing technieken?
Heb je ooit gehoord van de grote trefwoorden van sequencing technieken zoals "Next Generation Sequencing"? En hoe zit het met derde generatie sequencing? Als je antwoord niet echt goed is, ben je zeker op de juiste pagina beland!
In dit artikel leer je meer over de verschillende sequencing technologieën die er zijn en hun rol in het huidige biomedische onderzoek in het hart van het LUMC en andere onderzoeksinstituten over de hele wereld!
Deze gloednieuwe technologieën werden voor het eerst geïntroduceerd in de medische wereld tussen 2005 en 2006. Beginnend bij de eerste generatie technologieën - ook bekend als Sanger sequencing - ontwikkelden deze sequencing technologieën zich verder tot next generation sequencing (Illumina of MGI zijn enkele van de grote namen) en uiteindelijk tot de meest recente derde generatie technologieën die ons gebracht werden door de bedrijven Oxford Nanopore en PacBio.
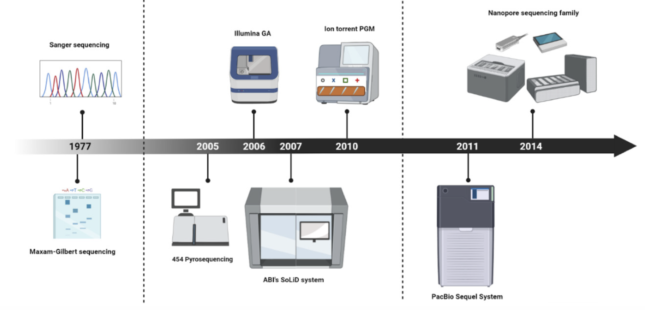
Vergelijking van methoden
Het belangrijkste verschil tussen deze sequencing technologieën is hun vermogen om stroken DNA van verschillende lengtes te lezen. Over het algemeen zijn eerste generatie technologieën in staat om korte DNA-fragmenten van ongeveer 100-600 basenparen per run te lezen, terwijl de volgende generatie sequencing op een veel nauwkeurigere manier kleinere reads (stukjes DNA die door het apparaat gelezen worden) van 50 tot 600 basenparen per keer detecteert. Derde generatie sequencing kan op indrukwekkende wijze meer dan 10.000 basenparen lezen, wat met andere woorden betekent dat het meer dan 10.000 DNA-moleculen in kaart kan brengen!
Het sequencen van DNA-fragmenten is belangrijk voor het blootleggen van genetische varianten die de oorzaak zijn van bepaalde ziekten bij individuen. We kunnen het zien als het barcoderen van een bepaalde variant die een ziekte kan veroorzaken, wat echt miljoenen mensen over de hele wereld kan helpen.
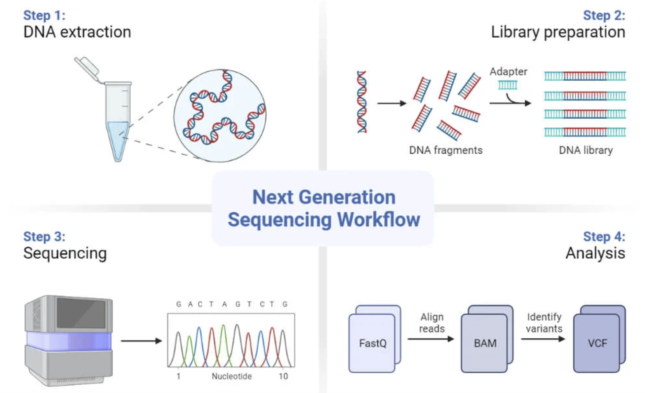
Stappen naar sequentiebepaling
Laten we eens dieper ingaan op hoe deze technologieën eigenlijk werken: er zijn vier belangrijke sequentiestappen. Allereerst moeten de DNA-monsters worden voorbereid en hierbij wordt het DNA uit een monster gehaald. Tijdens deze stap wordt het DNA gezuiverd van alle eiwitten of transcriptiefactoren die er nog aan gekoppeld zouden kunnen zijn. Tijdens elk van deze stappen zorgt de wetenschapper ervoor dat er kwaliteitscontrolemaatregelen worden genomen om eventuele afwijkingen in de resultaten te kunnen opsporen.
Ten tweede moet er voorafgaand aan het sequentieproces een zogenaamde bibliotheek worden voorbereid. Hier bereiden we ons DNA voor zodat het compatibel is met de sequencer. Dit wordt gedaan door het DNA te in kleinere stukjes te hakken waaraan we aan elk uiteinde specifieke adapters toevoegen. Het DNA wordt vervolgens vermenigvuldigd zodat we veel kopieën van specifieke DNA-regio's hebben; dit zal de nauwkeurigheid van onze uiteindelijke lezing van het DNA-sjabloon vergroten nadat de sequencing is uitgevoerd.
De derde stap is het eigenlijke sequencen, dat in cycli gebeurt. Tijdens elke cyclus wordt één nucleotide per keer opgenomen in het DNA dat zichzelf weer opbouwt, net als bij een legotoren. De strengen die we in de vorige stap hadden vermeerderd, fungeren als de blauwdrukken die het DNA nodig heeft om te weten welke nucleotiden het moet toevoegen. Elke nucleotide (A voor arginine, C voor cytosine, T voor tyrosine en G voor guanine) heeft een andere kleur en een ander signaal dat door het apparaat kan worden gedetecteerd.
De laatste stap is de analyse van de DNA-uitlezingen die aankomen in een FastQ bestandsformaat. Samengevat is deze stap waar we de korte DNA-lezingen samenvoegen om een groter beeld te krijgen van wat er gebeurt. De post-processing bestaat uit het uitlijnen van de reads met een referentie DNA genoom, dit is de informatie die in BAM bestanden zit. Uiteindelijk wordt deze informatie opgeslagen in VCF-bestanden zodra er varianten zijn geïdentificeerd.
Korte vs. lange sequenties
Interessant genoeg werkt sequencing van de derde generatie op een iets andere manier dan de eerste twee sequentiemethoden. In plaats van onderzoekers te voorzien van korte DNA-reads, levert het lange DNA-reads. Dit is extreem nuttig als we een gebeurtenis willen identificeren die bijvoorbeeld meerdere naburige varianten met elkaar kan verbinden. We kunnen het ons voorstellen als de mogelijkheid om uit te zoomen op een variant en te zien of er regio's zijn die mogelijk ook een bepaalde ziekte veroorzaken. Dit was een grote doorbraak aangezien RNA wordt afgebroken, wat voor korte reads zorgt. Voor langere reads heb je dus een delicate sequencer nodig.
Het is belangrijk om op te merken dat deze technologieën evenveel voor- als nadelen hebben. Hoewel ze onderzoekers een extreem hoge verwerkingscapaciteit bieden, wat betekent dat ze op een kostenefficiënte manier veel monsters tegelijk kunnen sequencen, kan hun output gemakkelijk erg complex worden om te analyseren en is er geavanceerde bioinformatica-expertise nodig om ze te interpreteren. De meest innovatieve machines hebben de tijd die ze nodig hebben voor elke run aanzienlijk verminderd en hebben een veel hogere nauwkeurigheid dan de eerste technologieën die werden ontwikkeld, maar ondanks deze vooruitgang is er nog steeds een groot aantal fouten. De nieuwe opkomende uitdaging voor deze technieken is het vergroten van de lengte van de gelezen data per run, zoals eerder beschreven. Het verkrijgen van langere lezingen van het DNA zou een meer zorgvuldige herbouw van genoomsequenties mogelijk maken.
Relevantie van deze technologieën
De manier waarop het huidige onderzoek functioneert is dat wetenschappers gespecialiseerd zijn in hun vakgebied, wat betekent dat ze bijvoorbeeld leren om de ene sequencingtechnologie te gebruiken in plaats van de andere. Deze onderzoeksfocus kan de analyse en diagnose van bepaalde ziekten, zoals kanker of neurologische ziekten, enorm helpen. Shortread-methoden zoals Illumina zijn bijvoorbeeld ideaal om kleine genomen of gerichte delen van het DNA te identificeren. Aan de andere kant zijn longread-technologieën zoals PacZBio of Oxford Nanopore sequencing briljant in het ontrafelen van sommige van de meer complexe structurele variaties in DNA-gebieden. Een combinatie van deze verschillende sequentiebenaderingen kan veel meer informatie opleveren dan wat in eerste instantie werd gevonden, wat onderzoekers tot uitgebreidere resultaten en interpretaties kan leiden. In sommige gevallen kan het zelfs de diagnose van een zeldzame ziekte verbeteren!
Ben je nog steeds nieuwsgierig naar deze technologieën? Lees dan de onderstaande bronnen of neem contact op met het Leiden Genome Technology Centre (LGTC) van het LUMC voor meer informatie over sequencing!
Bronnen:
Aryal, S. (2022, August 5). Next-Generation Sequencing (NGS)- Definition, Types. Microbe Notes. https://microbenotes.com/next-generation-sequencing-ngs/
Cuber, P., Chooneea, D., Geeves, C., Salatino, S., Creedy, T. J., Griffin, C., Sivess, L., Barnes, I., Price, B., & Misra, R. (2023). Comparing the accuracy and efficiency of third generation sequencing technologies, Oxford Nanopore Technologies, and Pacific Biosciences, for DNA barcode sequencing applications. Ecological Genetics and Genomics, 28(100181), 100181. https://doi.org/10.1016/j.egg.2023.100181
NGS Workflow Steps. Illumina.Com https://emea.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-workflow.html
How nanopore sequencing works. (n.d.). Oxford Nanopore Technologies https://nanoporetech.com/platform/technology
Leiden Genome Technology Center https://www.lumc.nl/en/research/facilities/leiden-genome-technology-center-lgtc/
Athanasopoulou, K., Boti, M. A., Adamopoulos, P. G., Skourou, P. C., & Scorilas, A. (2021). Third-generation sequencing: The spearhead towards the radical transformation of modern genomics. Life (Basel, Switzerland), 12(1), 30. https://doi.org/10.3390/life12010030
Lee, H., Gurtowski, J., Yoo, S., Nattestad, M., Marcus, S., Goodwin, S., Richard McCombie, W., & Schatz, M. C. (2016). Third-generation sequencing and the future of genomics. In bioRxiv. https://doi.org/10.1101/048603
0 Reacties
Geef een reactie